Information Research
Vol. 29 No. 2 2024
Metadata functional requirements for genomic data practice and curation
DOI: https://doi.org/10.47989/ir292363
Abstract
Introduction. The rapid accumulation of genomic data and their widespread reuse in clinical and scientific practice demands more effective description and organization of genomic information, which poses new challenges in developing metadata schemas. Based on a previously developed taxonomy of metadata requirements, this paper reports the results from a survey that addresses these new challenges.
Method. The survey gathered empirical data from 156 genomics scientists to identify context-sensitive metadata functional models for genome curation. The study further investigated the metadata elements from four well-known genomic metadata schemes against the functional requirements for genome metadata.
Analysis. The survey data were employed the statistical package STATA, to produce descriptive statistics, factor analysis, Fisher’s exact test, and related reports.
Results. Analysis of the empirical results revealed that genomics scientists recognize specific sets of criteria for metadata needs in the genome-curation context. Twenty one metadata requirements were reduced to five factor constructs. The ranking of these constructs in decreasing order is: portability, reusability, manipulability, sufficiency, interoperability, extensibility, and modularity. The Fisher exact test results revealed that the genomic community required rich context and technical related metadata elements to facilitate data exchanges and experimental operations in genome curation.
Conclusion. The findings indicated that genomics scientists developed metadata to meet the needs in genome curation activities related to data wrangling, integrations across platforms and databases, and data reuse. Architectural layout as flat file needs extra administrative metadata to support data sharing and documentation. The resulting metadata requirement model can serve as valuable resources to genome scientists, data curators and administrators for designing metadata schema and developing data-curation policies.
Introduction
Advances in computational tools and biological knowledge have enabled large-scale production of genomic information in last three decades and created ‘genomic Big Data’ Pruitt et al., 2012; Stathias et al., 2018). While advances of sequencing technologies propelled rapid growth in genomic data and brought many unique benefits to life sciences, they also created vast genomic data silos. The use of diverse computational tools, lack of awareness of data curation best practices, and lack of consensus on data management standards and practices are among the primary factors responsible for these data silos (Paten et al., 2015; Wilkinson et al., 2016). Just as metadata description practices vary across life sciences communities, practice inconsistencies exist even within the data produced through genomics curation activities (Chen et al., 2011; Yilmaz et al., 2011). As such, genomic data can be brittle and unfeasible for discovery, reuse, and preservation, creating barriers for realizing the value and full potential of genomic data. Researchers have called for standardization of curation procedures, metadata, data quality models, and tools to support facilitation of documentation, exchange, archival procedures, and reuse of data within the genomic research community (Griffin et al., 2017; Huang H., 2018; Lee, Kibbe, and Grossman, 2018; Klimke et al., 2011). These are critical for making data findable, accessible, interoperable, and reusable (FAIR) (Wilkinson et al., 2016).
The complexity in genomic data and related yet disparate information resources, however, poses great challenges in developing metadata schemes or infrastructures that are both easy to use and effective in facilitating genomic data documentation, exchange, archiving, and reuse (Paten et al., 2015, Kottmann et al., 2008; Liolios et al., 2009; Deck et al., 20 integrated genomic sequence data, experiments, and literatures (Pruitt et al., 2012). Genomic data includes genome sequence information and functional regions for gene transcription, the process of protein translation from gene, gene expression data, and related biological analysis (Crick, 1970). 09). Genome curation involves multi-faceted activities such as data use at different levels thatUsing state-of-the-art experimental methods, scientists generate, collect, and use genomic data to develop theories, models, and to perform integrative analysis (Stathias et al., 2018; Chen et al., 2011; Hong et al., 2016). These data are available in a variety of formats and organized and curated, but the quality of curation largely relies on the metadata schemas and tools that support the discovery, selection, retrieval, evaluation, and analysis of genomic data (Rapp and Wheeler, 2005). While metadata schemas and data management tools exist for genomic data curation and preservation (Klimke et al., 2011; Kottmann et al., 2008), they share a minimum consensus on certain data elements requirements, and are typically done on an ad hoc basis. In addition, metadata schemas are rarely evaluated and validated with empirical evidence. It is not uncommon that, when a project adopts a metadata standard, it has to be modified to fit the local description needs. In the process of modifying the standard(s), many elements are abandoned or left unused while many new elements have to be designed.
Genomic research involves diverse curation tasks and data practices, which implies the existence of multiple metadata schemes and tools (Cresto, 2002; Bernasconi et al., 2018). The long list of curation tools and metadata schemes provided in Klimke et al. (2011) are good examples. Some of the schemes in the list of Klimke et al. are incomplete or very specific for local databases. Constant updates have to be performed to add new metadata elements or drop those that are out of date. This further complicates the problem because the metadata descriptions that are already created under the old metadata schemas need to be updated or they will face the risk of being out-of-date or completely lost. The problem can become even worse with large scale sharing of data.
Understanding the functional requirements scientists expect for metadata standards and preferences they have for different types of metadata is essential for building better information infrastructures and systems to accommodate genomics curation needs, because clearly defined metadata functional goals can help guide the community in developing domain-feasible metadata schemas for genome curation. Questions that need to be addressed in the process of identifying the functional requirements for genomic metadata standards include: How do genomic scientists prioritize metadata functional requirements? What motivates them to develop metadata schemas? Are there any underlying similarities or trends in the way scientists develop metadata to facilitate their curation work? Answering these questions will enhance our understanding of scientists’ practices and preferences in developing and using metadata schemas, which in turn can help shape metadata standards that more closely adhere to the needs for resource discovery, data analysis, and data sharing across the existing genomics databases.
This paper reports the findings from a survey designed to understand metadata functional goals in genome curation. In addition, it surveys the available metadata standards and their metadata elements and makes comparisons.
Literature Review
Metadata Functional Requirements
Metadata as ‘structured data about data’ (Miller, 1998; Duval et al., 2002) has received a wide recognition for its role in managing data and facilitating data discovery and long-term curation. The attributes captured in metadata descriptions address questions such as what an object is and is about, who created it and when, and what technical requirements there are in order to read or display the content, be it a document, an image, or reusable information-bearing entity (Garshol, 2004). These functionalities place metadata in a pivotal role in information systems, especially those repositories and infrastructures designed to manage and organize research data (Greenberg, 2010; Huang and Jorgensen, 2013) and provide discovery and re-use services, as well as support searching and access to the resources (Duval et al., 2002; Garshol, 2004). Based on the functions of metadata, they are typically categorized as descriptive, administrative, technical, and structural types (Caplan, 2003; Zeng and Qin, 2022).
In the FAIR (findable, accessible, interoperable, reusable) principles, metadata is raised to a high level of importance because of its role across all four areas (Wilkinson et al., 2016; Boeckhout et al., 2018; Corpas et al., 2018). When metadata functionalities are expanded to other types of scientific research data, such as genomics data, understanding metadata functional requirements is to learn how to use metadata in genomics data for practical and useful purposes when conducting genomics curation activities and tasks. And it is related to how metadata works or operates in genomics curation work.
Using ontologies to model metadata schema has become a common practice in the metadata community in the last two decades, including biomedical metadata. The well-known BioPortal (https://bioportal.bioontology.org/) is claimed to be the world's most comprehensive repository of biomedical ontologies. These ontologies define biomedical entities and the relationships between these entities in machine-processable formats, which creates the necessary condition for developing software applications (Whetzel et al., 2011). Although the primary goal of these ontologies is not for conventional metadata schema development, many of them can serve as metadata models for curating genomic and other biomedical data and building knowledge graphs by utilizing the semantic web technologies. Among the many ontology-based metadata projects is the Ontology for Biomedical Investigations created as an ontology-based metadata model to the Biomedical community (Peters, 2009; Bandrowski et al., 2016). The Ontology for Biomedical Investigations provides additional genomic project-specific metadata elements, as well as ontology-driven metadata enrichment in genomic research (Dugan et al., 2014; Bernasconi et al., 2018).
The idiosyncrasies of scientific data in different disciplines require metadata descriptions to meet the functional needs in supporting the management, long-term curation, discoverability, and reusability of scientific data. Metadata standards have been used as a source to examine what functionalities are covered and how they can be generalized as guidelines for developing new metadata schemas for scientific data descriptions (Yilmaz et al., 2011; Qin et al., 2012). Informed by the Functional Requirements for Bibliographic Records (FRBR) task model, Willis and Greenberg (2012) conceptualized ten user-functional and architectural tasks that can support scientific data preservation and discovery. Major functionalities such as resource discovery and use, data interoperability, automatic metadata generation, linking publications and underlying datasets, data quality control and data security have been discussed (Qin et al., 2012). Based on Greenberg’s (2005) metadata objectives, principles, domains, and architectural layout (MODAL) framework, Willis et al. (2012) identified twenty-two metadata-related goals in multiple scientific domains from existing content and literature. This is a comprehensive list of widely applicable metadata goals and requirements in support of data sharing across disciplines and domains for scientific data management, dealing with data intensive applications.
Domain experts, researchers, and cataloguers have taken on the important role of developing metadata elements for scientific data managements (Qin et al., 2012). Indexers and cataloguers were found to have different levels of accuracy preferences for Dublin Core elements (Greenberg et al., 2006), while particular metadata elements can impact application usability and metadata curation (Crystal and Greenberg, 2005; Greenberg and Robertson, 2002).
Metadata elements can be grouped roughly into two types based on the metadata’s functionality for resource discovery and data administration: intrinsic (i.e., those that are related to resource identification and discovery) and extrinsic (i.e., those that are related administration and other non-bibliographic data) (Burnett, Ng and Park (1999). When considering metadata for a particular domain, metadata types may also be categorised as domain-specific, domain-independent, physical, and user metadata (Singh, et al., 2003). Domain-specific metadata can be very specific to a work domain (Ouzounis and Karp, 2002), for example, describing application data in different scales within the domain; supporting reconciliation of domain-specific data descriptions; and providing a set of horizontal mappings of data elements across disparate databases (Ouzounis and Karp, 2002). Domain-independent metadata, however, refers to those general descriptions such as ‘the creator/modifier of data and authorization/audit/lineage information related to the data’ (Ouzounis and Karp, 2002).
Metadata and genome curation
Research on genomes of humans and other living organisms has benefitted from low-cost, high-capacity sequencing technologies. The approach of sequence first, think later generated and is still generating a flood of genomic data that requires effective organization and analysis. Tremendous efforts have been made to build up information repositories and metadata schemes that include important genome features using the “gold standard” with experimental evidence (Ouzounis and Karp, 2002). The effort of providing open access and community-based data curation to genomic data while integrating data from different contexts highlights the need for metadata development, especially the need for aiding data standards development and data reuse based on genomic data characteristics and scales within the genomic research community. The genomic communities identified developing curation procedures, data quality assurance strategies, and supporting annotation tools as priorities in order to fully capture and validate the source metadata (Cochrane et al., 2007).The related data curation may evolve and change over time as the amount of data grows and more attributes need to be included in metadata schemas.
Metadata development and related issues in genome curation work are extensively discussed in literature (Liolios et al., 2009; Crasto et al., 2002; Barrett et al., 2011, Caufield et al., 2018). Genomic metadata plays the role of facilitating the description of genomic raw sequences, genetic code variations, and gene expression and function (Cochrane et al., 2007). Contextual metadata captures geographic and habitat attributes such as location and time that describe where and when a gene or organism was sampled (Chen et al., 2011; Chervitz et al., 2011; Hankeln et al., 2011, Wilkinson et al., 2016). This type of data is a prerequisite to understand gene functions and relationships between host and environmental conditions for biodiversity, evolutionary biology, and environmental health studies (Chen and Sarkar, 2011; Cochrane et al., 2007; Hankeln et al., 2011).
Conventionally, metadata schemas (a collection of metadata elements with certain relationships) for genome curation capture information about experimental description and execution, data reporting and exchange, and terminology or ontology (Chervitz, et al., 2011). For genomic experiment related metadata schemas, one focus is to capture the information about sample sources, such as sample locations, date and time of sampling, and environmental or ecological variables that can be attached with the genome sequences (Wilkinson et al., 2016; Chen and Sarkar, 2011; Hankeln et al., 2011). In addition, genomic metadata schemas must provide minimum information exchanges to support data reporting (Field et al., 2008). The Genomic Standards Consortium, an open-membership international organization formed in 2005, published the Minimum Information about a Genome Sequence specification to define a set of core (required) metadata elements for genomes that are needed to ensure that submitted data are sufficient for interpretation and retrieval by other scientists (Field et al., 2008). Another major metadata schema in the biological sciences is Darwin Core, a standard for describing biodiversity and biological specimens and promoting consistent use of a core vocabulary to describe taxa as documented by observations, specimens, and samples (Wieczorek, 2012). Genomic databases such as that of the National Center for Biotechnology Information and Ensembl (of the European Bioinformatics Institute) offer integrated, extensible, and re-usable metadata infrastructure for generating, storing, retrieving, and displaying genomic annotation data (Barrett et al., 2011).
Standardized computer markup languages are also used for encoding genomic data and documents. The Genomic Standards Consortium implements the Minimum Information about a Genome Sequence in XML as Genomic Contextual Data Markup Language, and specifies the use of persistent identifiers (e.g., PubMed identifier, digital object identifier), controlled vocabularies, and ontologies (e.g., the Environment Ontology) for most genomic metadata in the standard (Kottmann, et al., 2008; Field et al., 2008). As semantic Web technologies become widely accepted, libraries, institutions, governments, and communities have created generalized data repositories, disseminating data and the reference sources used in entity resolution as linked data (e.g., DBpedia; LinkingOpenData) through open access and use as biological resources (Zappa et al., 2012). Examples of such include the development of the National Center for Biomedical Ontology and the Gene Ontology (Camon et al., 2004; Smith et al., 2007; Jupp et al., 2014).
Methods
The primary focus of this exploratory study was to understand the perceptions of genomics scientists regarding metadata requirements in genome curation. In particular, the study was designed to address the following research questions:
RQ1: What are the metadata criteria considered to be important in genome curation work?
RQ2: How can existing genomic metadata schemas be characterized for their use in genomic data curation?
The first question was designed to gather information for determining the priority rankings and factor constructs of metadata requirements, which would serve as the metadata functional requirement model for genome curation. The second question addresses the differences in existing genomic metadata schemas by comparing and contrasting the metadata elements.
This study includes two parts: an online survey to collect genome scientists’ perceptions and opinions about genome metadata functions and a comparative study of metadata schemas based on the survey findings.
Survey design, recruitment, and data analysis
The study collected and analysed survey data. The survey questions were collected and modified from the previous twenty-two metadata functional requirements items found in the literature (Willis et al., 2012). In order to provide a context for the questions, the survey used a representative scenario to conceptualize activities relevant to the metadata functional requirement in genomic data practice and curation. The scenario was developed by using scenario-based task analysis (Carroll, 1997; Diaper, 2004; Go and Carroll, 2004). The population for this analysis consisted of scientists conducting genomic research and familiar with metadata practice. The survey finally recruited 156 survey participants. They were genomics scientists who had published journal articles related to genomic metadata, curation and genomic research. A total of 4,012 authors (with their e-mail addresses) of genomic research papers related to genome annotation and metadata practice were collected from the PubMed database, 800 of which were randomly selected for inclusion in this analysis. Participants were given a scenario that represented and conceptualized genome metadata functional requirements activities. Participants were provided the same set of written metadata functional requirements for genome curation that can be used for understanding user perception. The Qualtrics software (http://www.qualtrics.com) was used to distribute the survey and collect data. Respondents were asked to rate the importance of metadata functional requirements on a seven-point Likert scale. The data were analyzed with STATA 16 software (https://www.stata.com/stata16/) to produce descriptive statistics, factor analysis, Fisher’s exact test, and related reports.
Comparative analysis
Four metadata schemes used in genomic communities for different purposes were compared in counting the number of metadata elements in each specific category (see Table 5). Table 5 demonstrates metadata elements represented in four broad classes of metadata schemes: genomic sequence annotation; experimental, terminology/ontology; and data report/exchange. Two criteria were applied in sample selection: 1) the scheme must be widely used in a genomic community; 2) the scheme must be used in an active scientific data repository.
Two researchers coded the sampled metadata elements independently. For good qualitative reliability, Miles and Huberman (1994) recommended that the consistency of the coding be in agreement at least 80% of the time. To establish inter-rater reliability, two researchers independently coded metadata elements, which resulted in 92% inter-rater agreement. The categories and subcategories that emerged from the data are summarized and illustrated in Table 6.
Findings
Survey participants’ characteristics
Survey participants self-identified their curation roles as follows: end users (n=104, 67%), curators (n=52, 33%). 84% (n=131) of the participants indicated that they had experience with applying metadata or standard vocabularies in their research work. Over half of them (n=88, 58%) had a biology background, worked in higher education in the U.S. or Canada (n=89, 57%), and held a doctorate (n=117, 75%) (see Table 1).
| Demographic category | n | |
| Curation role | ||
| End user | 104 (67%) | |
| Curator | 52 (33%) | |
| Disciplines | ||
| Biology | 88 (56%) | |
| Both | 40 (26%) | |
| Bioinformatics | 28 (18%) | |
| Residency | ||
| U.S. and Canada | 89 (57%) | |
| Europe | 30 (19%) | |
| Asia | 30 (19%) | |
| South America | 4 (3%) | |
| Oceania | 3 (2%) | |
| Africa | 1 (1%) | |
| Education level | ||
| Ph.D. | 117 (75%) | |
| M.S. | 27 (17%) | |
| B.S. | 12 (8%) | |
| Using standard vocabularies | ||
| Yes | 131 (84%) | |
| No | 25 (16%) | |
| Organization | ||
| University | 102 (74%) | |
| Industrial | 20 (14%) | |
| Government | 16 (12%) | |
| Nonprofit org | 11 (8%) | |
| Clinical practice | 3 (2%) | |
Table 1. Demographics of survey participants (n = 156).
Ranking of metadata requirements
The descriptive statistics of the metadata requirement rankings are given in Table 3. The mean, median, and standard deviation were calculated for each metadata requirement. On average, the participants ranked top five metadata requirements in the decreasing order as the following: data comparability, data portability, data retrieval, scheme simplicity, and data interchange. Data comparability was of the highest importance and abstraction the lowest, indicating that the curated genome data was expected to be highly heterogeneous and the related metadata elements need to be comparable when sharing data on a large scale in an open-access environment (Larsson and Sandberg, 2006; Oliver, 2006).
| Metadata requirements | # of responses | M | Mdn | Mode | SD |
| Data comparability: The scheme is intended to facilitate comparison of data sets. | 150 | 6.14 | 7 | 7 | 1.14 |
| Data portability: Data created using the scheme is intended to be “portable”—software application and operating system independent. | 153 | 5.92 | 6 | 7 | 1.30 |
| Data retrieval: The scheme is intended to facilitate the discovery and acquisition of data. | 148 | 5.76 | 6 | 6 | 1.32 |
| Scheme simplicity: The scheme is intended to be simple and easy to use. | 150 | 5.75 | 6 | 7 | 1.39 |
| Data interchange: The scheme is intended to facilitate data interchange among community members—also referred to as data exchange, data sharing, or data communication. | 147 | 5.73 | 6 | 7 | 1.27 |
| Data publication: The scheme is intended to support publication of data in journals and databases. | 147 | 5.46 | 6 | 6 | 1.41 |
| Data validation: The scheme is intended to facilitate validation of data through the use of strongly typed data values. | 145 | 5.39 | 6 | 6 | 1.46 |
| Data documentation: The scheme is intended to describe not only the data, but to document the data context (experimental or observational context, analytical methods, etc.). | 146 | 5.38 | 6 | 6 | 1.35 |
| Data archiving: The scheme is intended to facilitate the preservation/archiving of data sets and data documentation. | 146 | 5.34 | 6 | 6 | 1.37 |
| Scheme simplicity: The scheme is intended to be simple and easy to use. | 144 | 5.30 | 6 | 6 | 1.39 |
| Sufficiency (minimal set): The scheme defines the minimal amount of information needed to achieve a specific goal for the community, for example, secondary data reuse (e.g., DDI, EML), experiment verification/reproduction (e.g., MINiML). | 145 | 5.25 | 5 | 6 | 1.48 |
| Technical stability: The scheme implementation will not change, will be supported over time, and is safe to adopt. | 148 | 5.23 | 5 | 6 | 1.41 |
| Provenance: The scheme is intended to document the origin of information. This includes the origin of the data set (e.g., EML, DDI ) or the origin of elements in the data set (e.g., ThermoML, mmCIF ). | 145 | 5.21 | 5 | 5 | 1.27 |
| Inter-scheme modularity: Elements from the scheme are intended to be used in conjunction with elements from other schemes (19) to meet new purposes. | 146 | 5.16 | 5 | 6 | 1.44 |
| Conceptual stability: Concepts represented in the scheme are stable and will not change over time. | 149 | 5.07 | 5 | 5 | 1.47 |
| Data lifecycle: The scheme is intended to support documentation of the data lifecycle—changes that occur to the data set over time. | 149 | 5.01 | 5 | 5 | 1.41 |
| Scheme harmonization: The scheme is intended to be compatible and interoperable with other related schemes (e.g., DDI, EML) or the scheme was derived from an existing scheme (e.g., Darwin Core/Dublin Core, mmCIF/PDB). | 144 | 4.98 | 5 | 5 | 1.40 |
| Element refinement: Element refinement is the ability to make more specific the meaning of an element (19). This is achieved through type extension (subclassing, deriving, subtyping). Refined elements can still be used in standards-based systems. | 143 | 4.82 | 5 | 5 | 1.36 |
| Comprehensiveness: The scheme is intended to provide a comprehensive set of elements (or vocabulary) to describe a particular aspect of the domain. This is generally indicated by phrases such as “cover all” or “encompass all.” For example, ThermoML is intended to “cover all experimentally determined thermodynamic and transport property data.” | 143 | 4.78 | 5 | 5 | 1.49 |
| Intra-scheme modularity : The scheme itself is modular and intended to support use of subsets of elements (or modules) for a particular purpose or particular stage of metadata creation. Modularity may also mean that data can be stored in multiple files or assembled at different times (DDI ). | 144 | 4.76 | 5 | 5 | 1.36 |
| Core set: The scheme is intended to provide only a core vocabulary, a common set of elements used to describe the most common situations (e.g., Darwin Core). | 148 | 4.75 | 5 | 5 | 1.64 |
| Scheme flexibility: The scheme is intended to be adapted for use in settings outside of the current context. | 145 | 4.73 | 5 | 5 | 1.43 |
| Abstraction: A conceptual model has been defined and is intended to be separate from the particular technical implementation. | 142 | 4.34 | 5 | 5 | 1.48 |
Table 2. Survey participants’ ranking of metadata requirements by mean importance in the context of Scenario.
Factor constructs for metadata requirements
To identify the major factor constructs for the twenty-two items of metadata requirements reflected by the 153 survey respondents’ rankings, an exploratory factor analysis was employed using principal component analysis as the extraction method and varimax with Kaiser normalization as the rotation method (see Table 3). The cut-off size for criterion loadings was set to 0.45 based on the sample size n=153 (53). Both the Bartlett (X2 = 1102.19, p < 0.001) and measure of sampling adequacy (MSA = 0.805) tests for the sample indicated a significant level of correlation among the metadata requirements. A scree-plot analysis suggested selecting the first six components for metadata requirement constructs.
| Components | ||||||
| Metadata requirements | 1 | 2 | 3 | 4 | 5 | 6 |
| Abstraction | 0.15 | -0.17 | 0.51 | 0.16 | 0.60* | 0.21 |
| Comprehensiveness | 0.28 | 0.38 | 0.54* | 0.12 | 0.07 | -0.13 |
| Conceptual stability | 0.17 | -0.17 | 0.29 | 0.72* | 0.12 | 0.24 |
| Core set | -0.14 | 0.15 | 0.01 | 0.06 | 0.07 | 0.80* |
| Data archiving | 0.78* | 0.05 | 0.24 | 0.17 | -0.08 | 0.03 |
| Data comparability | 0.34 | 0.62* | -0.18 | 0.31 | 0.28 | 0.03 |
| Data documentation | 0.65* | 0.17 | 0.39 | -0.01 | 0.11 | 0.15 |
| Data interchange | 0.78* | 0.20 | -0.03 | 0.25 | 0.22 | -0.04 |
| Data lifecycle | 0.52* | -0.09 | 0.03 | 0.17 | 0.37 | 0.22 |
| Data portability | 0.24 | 0.23 | 0.04 | 0.54* | 0.29 | -0.19 |
| Data publication | 0.70* | 0.12 | 0.24 | 0.19 | 0.11 | -0.06 |
| Data retrieval | 0.64* | 0.42 | 0.09 | -0.05 | 0.19 | -0.15 |
| Data validation | 0.10 | 0.62* | 0.20 | 0.10 | 0.04 | 0.26 |
| Element refinement | 0.19 | 0.59* | 0.36 | 0.18 | 0.17 | 0.01 |
| Inter-scheme modularity | 0.21 | 0.35 | 0.05 | 0.08 | 0.70* | 0.02 |
| Intra-scheme modularity | 0.08 | 0.35 | 0.31 | 0.08 | 0.62* | -0.06 |
| Provenance | 0.27 | 0.06 | 0.64* | 0.01 | -0.05 | 0.30 |
| Scheme extensibility | -0.01 | 0.44 | 0.60* | 0.13 | 0.22 | -0.16 |
| Scheme flexibility | 0.19 | 0.12 | 0.66* | 0.08 | 0.40 | -0.05 |
| Scheme harmonization | 0.10 | 0.71* | 0.17 | 0.07 | 0.20 | 0.28 |
| Scheme simplicity | 0.00 | 0.45 | 0.01 | 0.73* | -0.09 | -0.09 |
| Sufficiency (minimal set) | 0.35 | 0.44 | 0.03 | 0.06 | -0.09 | 0.58* |
| Technical stability | 0.29 | 0.18 | 0.04 | 0.52* | 0.11 | 0.25 |
Table 3. Factor loadings for the metadata functional requirements.
The metadata requirements loaded on the first factor construct could be categorized as those of Reusability (see Table 4). The second construct was mainly related to Interoperability. The third was about Extensibility. The fourth construct included portability, which had the highest loading. The fifth construct was modularity. The criteria loaded on the sixth construct could be categorized as related to sufficiency. The constructs were then ranked by the arithmetic averages of the mean ratings of the metadata requirements loaded on the constructs (see Table 4). The portability construct was ranked the highest, followed by the reusability, interoperability, sufficiency, extensibility, and modularity constructs (Table 4, Figure 1).
|
Avg | Metadata requirements |
|
5.49 | Conceptual stability, data portability, Scheme simplicity, Technical stability |
|
5.45 | Data lifecycle, Data archiving, Data publication, Data interchange, Data retrieval, Data documentation |
|
5.33 | Data comparability, Element refinement, Scheme harmonization, Data validation |
|
5.00 | Core set, Sufficiency (minimal set) |
|
4.87 | Comprehensiveness, Provenance, Scheme extensibility, Scheme flexibility |
|
4.86 | Inter-scheme modularity, Abstraction, Intra-scheme modularity |
Table 4. The six factor constructs generated from the metadata requirements, and ranked by the arithmetic averages of the mean ratings of the individual requirements loaded on the constructs.
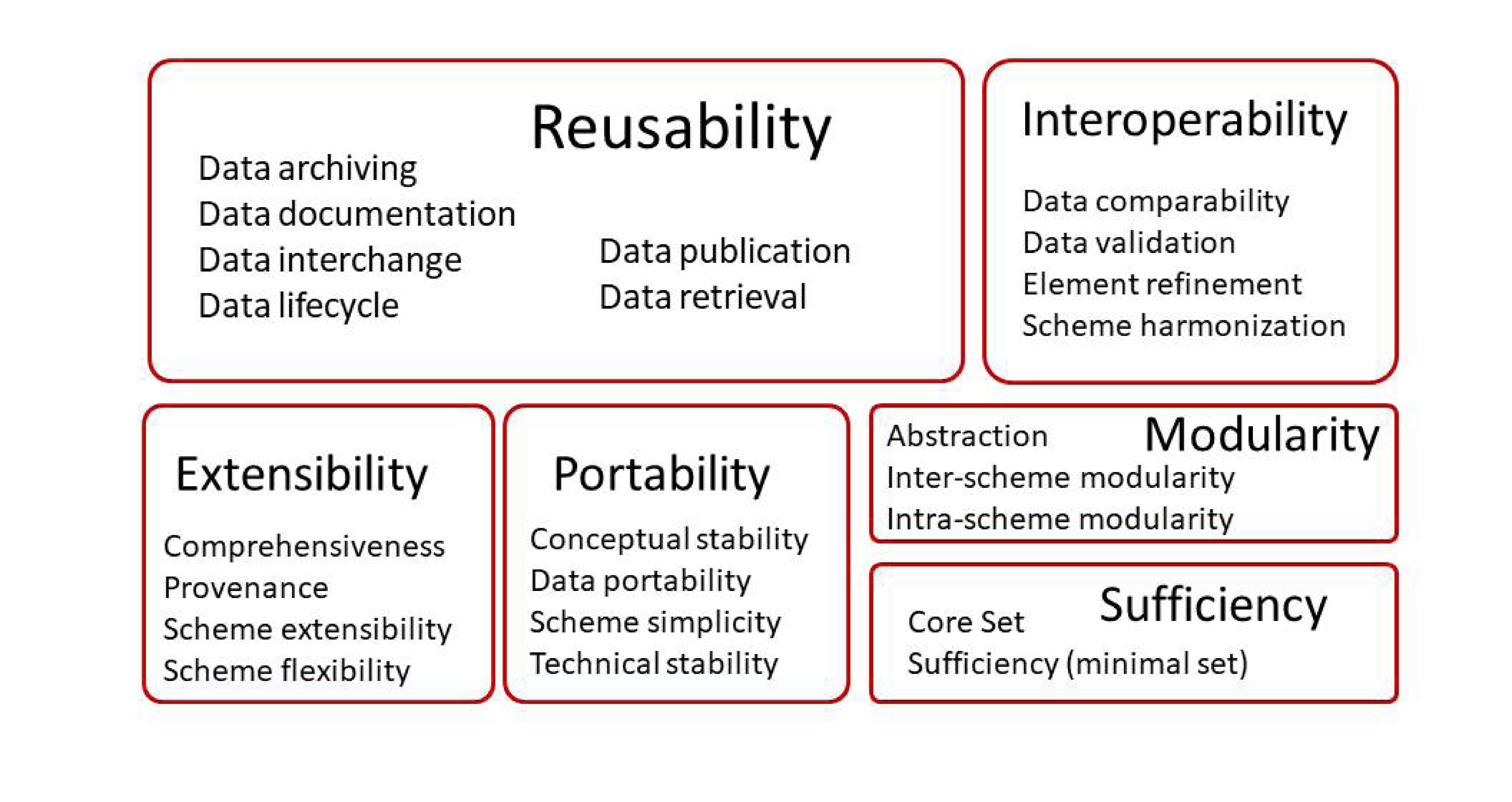
Figure 1. Metadata model in genome curation work
Comparison of available genomic metadata schemes
Among the many different ways of grouping types of metadata as reviewed in section 2, we decided to adopt the architectural and functional perspectives for analysing the data set for this project (Qin et al., 2012; Qin and Li, 2013). In this study, metadata elements from selected genomic metadata schemes were categorised based on three main functional views: resource identification and discovery, scientific context, and administration (Table 6). Metadata elements used for resource identification and discovery are the terms, ontologies, and attributes that describe and annotate resources for resource identification and discovery. Metadata elements describing scientific contexts are those used to represent standardised workflows, procedures, and project protocols related to the particular genomic studies or projects. Finally, administrative metadata document the information about metadata records such as metadata structures and elements.
| Functional and Architectural view | Related elements | Definition |
Resource identification and discovery: metadata about work agent, investigation, publication, and data set or collection
|
Identity | • The name of an entity that is used to identify the entity understood by human users. • A unique ID either in the form of some code or of a string following an identification system. |
| Descriptive | • General attributes about what the resource is and when it is published, released, or made available. • Related resources of the resource that is described. |
|
| Semantic | • Subject terms describing the content of data. • Subject or classification categories. • Taxonomic classes. |
|
| Generic | • General-purpose elements, including comment, annotation, note, etc. | |
Scientific context: metadata about workflow, provenance, parameter, and processing
|
Context | • Information about study/project design, model, and population under study. • Data collection methods, instruments, and constraints. • Analysis methods used. |
| Technical | • Parameters, models, measurements used in the dataset. • Software-, system-, and format-related attributes. |
|
| Temporal | • Measurements of time. • Temporal coverage of the content of data. • Temporal criteria for data segmentation, processing. |
|
| Location | • Geographic names. • Geospatial coordinates. • Aerial maps and/or data. |
|
| Administrative: metadata related to administrative such as file size, storage medium and dissemination medium (for offline data) are typical examples | Administrative | • Information about metadata record, standard used, responsible party, rights for the metadata records. • Information about data archive/repository • Wrapper or nesting elements for structuring and syntactic purposes. |
Modified from Qin et al., 2012; Qin and Li, 2013.
Table 5. Metadata functional and architectural
categories used to analyse genomic metadata schemas
In Table 6, the data show noticeable variations in number for a particular metadata element type that serve for a typical data function and task. The metadata element type that occurred most frequently is descriptive metadata: it has the highest total number of occurrences for all four schemas. Another noticeable category is the context metadata for both experimental and report and data exchange standards (FuGE (https://fuge.sourceforge.net) and MINiML (https://www.ncbi.nlm.nih.gov/geo/info/MINiML.html)). The European Molecular Biology Laboratory (EMBL) scheme also has a number of administration-related elements that the other three do not have. FuGE shows the highest number of technical-related metadata elements when compared to the other schemes.
| Functional and Architectural view | Element types | EMBL flat file: sequence annotation | GO: terminology | FuGE: experiment | MINiML: report/data exchange | ||||
|---|---|---|---|---|---|---|---|---|---|
| Element examples | n | Element examples | n | Element examples | n | Element examples | n | ||
| Resource Identity and Discovery | Identity | Accession number, Identification, Project Identifier, |
3 | Accession, Name | 2 | URI, Identifiable |
3 | Accession, Public-iD, Sample-ID | 4 |
| Descriptive | Database cross-reference, Keyword | 11 | Version, dbxref | 5 | Database, Bibliographic Reference | 19 | Description, Ref Source | 15 | |
| Semantic | Organism, Classification, Organism species | 4 | Synonym, term, is_a | 9 | Ontology Individual, Material, Ontology term |
4 | Molecule Type, Relation, Organism Type | 5 | |
| Generic | Comments, Feature tables, | 2 | Comment, Definition, Association, File | 3 | 0 | Characteristics, Comment, Summary | 5 | ||
| Scientific context | Context | CDS (coding sequence), translation, | 2 | Evidence | 1 | Investigation, Protocol | 25 | Label-protocol, Manufacturer, Overall-Design | 20 |
| Technical | 0 | Qualifier | 1 | Parameter, Atomic Value |
23 | Channel-count, Tag-length | 6 | ||
| Temporal | Date, | 1 | Date | 1 | Audit | 1 | Last-update-date, Submission-Date | 2 | |
| Location | 0 | 0 | Address | 2 | 0 | ||||
| Administrative | Administrative | Assembly Header, Feature table, header Spacer line | 5 | 0 | 0 | 0 | |||
Table 6. Distributions of metadata elements of genomic metadata schemes in functional categories
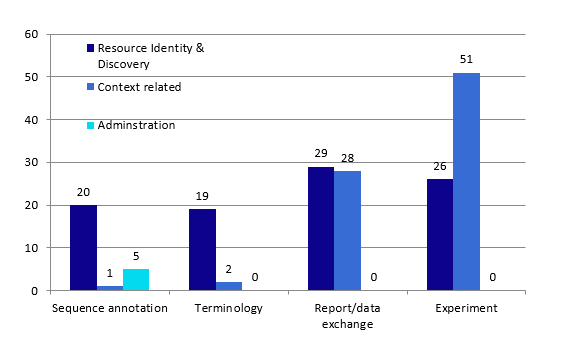
Figure 2. Four genomic metadata schemes and their three functional metadata categories
After aggregating the respective metadata elements intro three functional groups: resource identification, context related, and administration (as showed in Figure 2), all four metadata schemes contained a similar number of metadata elements for resource identification and discovery. Context related metadata elements in Experiment type metadata schemes had the highest number of metadata elements (n=51), followed by those in report and data exchange metadata schemes (n=28), terminology metadata schemes (n=2), and sequence annotation metadata schemes (n=1). A 2X2 contingency table Fisher’s exact test indicated that the occurrences of both resource identification and discovery and contextual metadata types in EMBL: sequence annotation and GO (https://www.geneontology.org): terminologies were significantly different from those in FuGE and MINiML. The number of occurrences of administration metadata in EMBL was also significantly different from the other three groups: GO, FuGE, and MINiML.
Discussion
This study reports for the first time the priority rankings of metadata types based on the feedback from genomic scientists. As the development and applications of genomic metadata evolves rapidly, the genomic community is becoming increasingly aware of the need for and value of metadata standards for genomic data sharing and curation. In addition, standards that become widely adopted can help scientists and data analysts better utilize, share, and archive the ever-growing mountain of genomic data sets.
The objectives of genome curation lie in marking up the key features of the genome and linking them to the related literature (Stein 2001). Genome curation is a collaborative activity, involving participation by many stakeholders from different domains (e.g., researchers, clinical doctors, and practitioners, especially those in institutional repositories) who might have different needs for and uses of the same information. For example, genome annotations link knowledge with specific gene products useful in the development of personalized genomic medicine. Genome-curation tasks include collecting raw genomic data and applying various tools for analysis of the primary data, i.e., utilizing available genomic information and secondary data for production of functional genomics interpretation and new knowledge for promotion of human health. Prior research indicated that researchers would like to obtain rich descriptive metadata supporting discovery and reuse, even in the absence of time and resources necessary to produce quality metadata (Greenberg et al., 2001). Metadata production in general is reported as inefficient, with automatic applications not being fully employed, and often the same metadata is being generated via humans in multiple settings (Greenberg, 2010).
Perception of genomic metadata functional requirements
The scientists ranked metadata portability the highest; this indicates that the quantity of genomic data exists on a large scale and that users need to access, compare and collect, and integrate disparate piece of scientific data sets across the databases (Schadt et al., 2010; Ding et al., 2010). Minimizing usability barriers is one of the main goals of metadata standard initiatives (Chervitz et al., 2011). Fostering the development of software tools that can help with file conversion to standardized representation further enhances utilization of standards during the research process (Chervitz et al., 2011).
What is proposed then, is that data schemes shall be independent and easily migratable between systems, while other metadata functional requirements cover the Portability construct. For example, conceptual stability indicated the concepts for genome curation represented in the scheme are expected to be consistently represented and stable. This is important for the technical solutions when metadata is recomputed and reparsed across the databases, and used to reduce the burden of technical challenges. Scheme simplicity indicated that scientists prefer metadata schemes as flat-structures and with not too many required data entry metadata elements. Metadata schemes with a fine-grained hierarchy and relationships among data elements might require a complicated data infrastructure that causes difficulty when migrating metadata from one collection to another. Complicated metadata schemes hinder metadata-compliant data sets for data sharing, and re-utilization. Therefore, implementation of metadata schemes could be technically stable when using software analysis tools to process data that conform to certain portable data standards. This also indicates that a simple form of metadata scheme both in format and hierarchy structure will benefit the community in various ways. When metadata schemes are in the form of complex and deep-layered structures, this makes automatic metadata generation extremely difficult (Qin and Li, 2013), attributable to differences between the types of information objects used by these two communities.
The reusability construct was ranked the second important in the genome curation metadata model. Within the construct, there were several metadata requirements that support data documentation and preservation (e.g., data life-cycle, data documentation, archiving). Metadata schemes improve both citation and discoverability of genome curation digital objects. Genome curation requires standards for data publication to recognize the value of data comparison and characterization (Chervitz et al., 2011). One of the objectives of metadata standardization is to ensure that metadata is constructed so as to prolong its longevity and accuracy (Chervitz et al., 2011). Updating and documenting changes (e.g., data life-cycle) in curation data allows curators to remain consistent, as well as ensures accuracy and currency of data curation practices (Vasilevsky et al., 2012). With metadata support to improve data traceability, it will enhance data interchange that facilitates data sharing, exchange, and communication. Data retrieval facilitated the discovery and acquisition of genomic data for future reuse.
As genomic data and other digital objects become larger and more diverse, and as analysis becomes increasingly more complex, it is necessary to develop an integrated and interoperable digital cyber-infrastructure that supports data sharing and communication. Interoperability constructs related to the metadata standard can be comparable with other schemes. The portability of the metadata standards relates to the ability in interoperable applications to reuse semantic elements in a metadata standard in different contexts (Qin and Li, 2013). Semantic elements in this study, are elements that describes the meaning of data. In certain genome curation tasks, scientists will have to integrate data from different sources which will require different metadata schemes. It will be good that these schemes can be compatible for harmonization. Equivalent or similarly-functioning metadata elements from two or more metadata schemes can be cross-walked, compared, and mapped to one another allowing detailed comparison among schemes (Greenberg, Spurgin, and Crystal, 2006).
However, the trade-off for harmonization is determining which metadata elements will be kept or discarded, since some valuable metadata will be missing during the cross-walking. For instance, it may be required to add more specific metadata to distinguish the respective digital object from others, or describe the specific and complex genomic data. Similarly, metadata may need to be refined in order to disambiguate two different item types representing structural (e.g., sub-classing, deriving, and subtyping) or semantic differences. Such processes can be facilitated by data validation to ensure the data type is compatible with the data type that had been defined. To facilitate the harmonization of terminologies or metadata schemes, innovative approaches are required to merge different metadata schemes, in order to dock different retrieval systems together, and propose a possible unified retrieval solution to obtain related genomic data and its annotation across different databases.
The sufficiency construct indicated that the genomic community requires core metadata elements for data entry of critical information stored in files. In order to obtain genomic curation data, the data must be accessible as well as the details of how the data minimal to adequately describe information objects (Chervitz, 2011). Standards for reporting, or minimum information, are needed to ensure that submitted data are sufficient for clear interpretation and querying by other scientists. Metadata related to sampling procedures and experiment verifications were fundamental to ensure data reproducibility or secondary use. The requirement for breadth-of-coverage of the metadata use depends on the necessity of the metadata to support secondary re-use (Qin and Li 2013). For example, metadata describing specific environments where the genome-sequencing biological samples were collected is a requirement for understanding the sample organism and their potential genome property. Some other data elements can be missing, not because they are not important, but simply because the genome curation community might keep certain metadata only for locally shared knowledge or self-checking purposes.
Extensibility is about the thoroughness and completeness of the metadata elements as it relates to comprehensiveness. It will be good for metadata standards that can maintain some degree of extensibility, even within a scientific domain like genomics, to have the potential to provide consistency and uniformity in the data generated by different interested parties (Chervitz et al., 2011). Scientists might care more about what had been updated and curated historically: in fact, they would like some flexibility and extensibility for metadata schemes so that they do not need to modify the scheme when the scheme is extended by adding new elements or module as update. This is a type of data quality trade-off as long as scientists can do backward tracing of modifications. Data provenance is referred to document the origin of information, and the documentation of the history of the annotation was reported to be very important (Bhardwak et al, 2014). Data provenance therefore facilitates effective reuse, integration, and analysis of that data to enable easier collaboration among different stakeholders. Provenance in this case is not just using metadata to record the origin of the data, but also to document the experimental workflow. The metadata is even used to document the process of data curation, transformation and derivation; and/or to capture actions that have an impact on data curation work (Davidson and Freire, 2008). By doing so, provenance can enhance the comprehensiveness of the curation pipelines or systems and optimize the curation process that allows users to better understand the curation mission and goals and increase compliance with curation policy or institutional mandates.
Lastly, the modularity construct indicated that genomic metadata schemes are required to support new metadata assembly based on established metadata schemas (Duval et al., 2002). Data elements from different schemes, as well as vocabularies and other building blocks, can be syntactically and/or semantically merged together to meet for new purposes (Duval et al., 2002). Subsets of data elements from a scheme can be easily reconstructed into a more fine-grained structure for special purposes (Duval et al., 2002). A variety of semantic modules can be mixed within a common syntactic foundation as a compound schema, so that can be represented using a common syntactic program language (e.g., XML) (Duval et al., 2002). This also enables the function of metadata abstraction, where a conceptual data infrastructure can be separate from the particular technical implementation. In this way, a modular set can be assembled to meet the specific local requirements of a given application without sacrificing too much interoperability when across domains (Duval et al., 2002).
Schemes and practices for genomic metadata
Analysis and cross-comparisons of current available metadata schemes and practices within the genomic community demonstrated the varieties of the specific requirements for metadata and scheme development. In general, the process of developing metadata and its applications began by establishing subgroups that represented various purposes and/or functions in genomics (Dugan et al., 2014). Each subgroup discussed and reviewed the internal or external resources of sequencing project and necessary metadata elements for the sample (Dugan et al., 2014). The comparison indicated that metadata that supporting resource discovery and description of contextual information are important for genomic research community. As found in the survey, a core set of metadata is expected in the genomics research community, the Genomic Standards Consortium (GSC) has defined a list of core (required) metadata elements, with the remaining as optional, for curation purpose (Field et al., 2008). The number of cross-references between core datasets, including metadata elements, has dramatically increased as researchers generate and exchange large data. Cross-reference from separate databases may cause data redundancy and inconsistency problems (Goll et al., 2010; Klimke et al., 2011). Given the non-static nature of the genomics field, new knowledge and concept appear daily, and their old curation links might be invalid if not updated timely (Khatri et al., 2005; Goll et al., 2010). Understanding the metadata functional requirements will help develop strategies to meet the local needs and support resource discovery, analysis, use and reuse of existing data through repositories to address geographical, temporal and contextual changes in curated information. Data values, descriptions, and annotated terms were standardised with other controlled vocabularies, and prioritised based on their importance related to data access and analysis (Dugan et al., 2014). Finally, the project-level or sample-level metadata fields were integrated as one schema, while less important or redundant metadata elements were eliminated (Dugan et al., 2014).
While genomic research may have varying focuses, metadata schemes can be organized to facilitate sequence annotation, terminology, report and data exchange, and experiment (Chervitz et al., 2011). This study’s findings indicate that the structural design of a scheme, extent, and granularity of metadata elements are dependent on curation purposes. For example, an increase in the number of metadata elements related to contextual research design, protocol, and procedures were required for the schemes that support experimental data exchanges. This also has an impact on the selection of administrative metadata elements. Flat file designs lack granular building blocks that enable metadata description and annotation, so they require more administrative-related metadata elements to facilitate the compartments of specific metadata purpose.
As ‘standards are like toothbrushes’, people might just want to use their own metadata and scheme. The existing variations might be due to the unique metadata requirements from different subgroups of professional experts within the field. The genomic research community has experimented with metadata practices that are tailored to the needs of specific genomic research subgroups. What emerges is not a view of different standards to describe the same information or physical curation objects, but a rich set of curation tools which are uniquely designed to characterize a particular genomic data type.
Conclusion
Due to the field's rapid change with new research findings arriving in a daily basis, humongous amounts and diverse types of genomic related data continue to grow, necessitating new knowledge and information to be described by new metadata elements. Instead of a one-size-fits-all metadata solution to meet the needs of a variety of genomic related data, looking at metadata core functions and their use in genomics research context, and developing a set of metadata core functional requirements, can explore a context-aware coordinate or an evolving structure and path, that can guide researchers to continuously design and update metadata elements and their schemes, to efficiently conduct data exchange tasks, and build new knowledge in genomics research.
In general, metadata requirements are considered highly important in the genomic research community, and there are concerns about technical issues within metadata schemes such as portability, data element sharing/re-use, and documentation. Scientists would like to make sure that their data are preserved in a way that will be useful. The reusability construct is related to the data documentation support and data lifecycle, while interoperability was adapted for used within or outside the context. Scientists also believe that metadata can facilitate data reuse and data sharing. Metadata in the genomic community requires a simple format that supports a modularized, minimal core set of metadata. In the genomic community, the new metadata standards are all related to the minimal requirements of genome curation. Modularity indicated that the elements from the scheme were intended to be used for different purposes.
The metadata solutions could be scalable, flexible and capable of being re-contextualized for satisfying local or global sharing needs, and require effective data management of smaller scale studies. Data sharing mechanisms will not just deal with publications but also with real data and its metadata sharing. As a result, not all metadata schemes were designed by following community standards, but rather, they were targeted for local needs and vary greatly from scheme to scheme. This survey result suggests a change in the perception and practice that emphasizes data reusability and as such, the metadata is expected to support the function of data reuse. The resulting models can serve as valuable resources for genomics scientists and administrators to combine domain knowledge and knowledge representation in the development of metadata policies and schema in genome curation. The findings suggest the expectations respondents have toward the ideal metadata schemes and services, that is, they should facilitate genomic data comparisons, support data and metadata reuse. Thus, the effective, standardized genomic data curation process and strategies are needed for modelling and updating dynamic genomic entity metadata. Findings from this study will inform decisions about using standard terminology to transform metadata functional requirements into an actionable knowledge base: a web ontology machine readable language (e.g., OWL). On a practical level, results from this research could be used to develop guidelines for practitioners, aligning specific metadata requirements to improve genome curation. Understanding the functional roles of metadata or their purpose of metadata use and function in the genomic context, this paper emerged functional metadata categories that each comprises a specific metadata requirement dimension. Furthermore, scientists can consider these prioritization and structures of metadata requirements to develop new metadata mining tools by Artificial Intelligence or Machine Learning approaches for genomics research community.
This study also has some limitations. The data were collected by survey, rather than direct observation, to collect the opinions of the scientists regarding metadata requirements used to develop the data quality models for genome curation. The data are therefore only approximations of the respondents’ actual value models for metadata requirements in practice. Future research collection of additional empirical data through observations and interviews can help determine the community’s metadata practices. Additionally, the importance of these concepts was recorded by survey participants at the time of survey completion; the follow-up interview provides an opportunity to validate where modifications are necessary.
However, the findings from this study could provide incorporating feedback from the genomics community both at the standard development and on a continuing basis so that the standard can adapt to user needs that can change over time. The genomic research community does not expect having a single and rigid metadata schema, but an open and scalable metadata “ecosystem” to connect all genome curation objects with metadata as individual datasets, software tools, annotation pipelines and modules. Eventually, the goal of having such a metadata “ecosystem” is to embrace the curation changes or needs in a data driven research world.
Acknowledgements
The authors want to thank the three anonymous reviewers for their insightful comments.
About the authors
Dr. Hong Huang is Associate Professor at University of South Florida iSchool. His research areas encapsulate three related disciplines- information and library science, bioinformatics, and information and learning technology. His current research interests included data management, artificial intelligence and machine learning data practice and sharing, as well as genomics, learning and edutainment. He can be contacted at honghuang@usf.edu
Dr. Jian Qin is Professor of Library and Information Science at Syracuse University iSchool. She conducts research in areas of metadata, knowledge and data modelling, scientific communication, research collaboration networks, and research data management. Her research has received funding from IMLS to develop an eScience librarianship curriculum and from NSF for the Science Data Literacy project. She is the recipient of the 2020 Frederick G. Kilgour Award for Research in Library and Information Technology. She can be contacted at jqin@syr.edu
References
Bandrowski, A., Brinkman, R., Brochhausen, M., Brush, M. H., Bug, B., Chibucos, M. C., Clancy, K., Courtot, M., Derom, D., Dumontier, M., Fan, L., Fostel, J., Fragoso, G., Gibson, F., Gonzalez-Beltran, A., Haendel, M. A., He, Y., Heiskanen, M., Hernandez-Boussard, T., … Zheng, J. (2016). The ontology for biomedical investigations. PloS one, 11(4), e0154556. https://doi.org/10.1371/journal.pone.0154556
Barrett, T., Clark, K., Gevorgyan, R., Gorelenkov, V., Gribov, E., Karsch-Mizrachi, I., Kimelman, M., Pruitt, K. D., Resenchuk, S., Tatusova, T., Yaschenko, E., & Ostell, J. (2011). BioProject and BioSample databases at NCBI: facilitating capture and organization of metadata. Nucleic Acids Research, 40(D1), D57-D63. https://doi.org/10.1093/nar/gkr1163
Bernasconi, A., Canakoglu, A., Colombo, A., & Ceri, S. (2018). Ontology-driven metadata enrichment for genomic datasets. In 11th International Conference Semantic Web Applications and Tools for Life Sciences, SWAT4LS 2018 (Vol. 2275, pp. 1-10). CEUR-WS.
Bhardwaj, A., Bhattacherjee, S., Chavan, A., Deshpande, A., Elmore, A. J., Madden, S., & Parameswaran, A. G. (2014). Datahub: collaborative data science & dataset version management at scale. https://doi.org/10.48550/arXiv.1409.0798
Boeckhout, M., Zielhuis, G. A., & Bredenoord, A. L. (2018). The FAIR guiding principles for data stewardship: fair enough? European journal of human genetics, 26(7), 931. https://doi.org/10.1038/s41431-018-0160-0
Burnett, K., Ng, K. B., & Park, S. (1999). A comparison of the two traditions of metadata development. Journal of the American Society for Information Science, 50(13), 1209-1217. https://doi.org/10.1002/(SICI)1097-4571(1999)50:13<1209::AID-ASI6>3.0.CO;2-Y
Camon, E., Magrane, M., Barrell, D., Lee, V., Dimmer, E., Maslen, J., Binns, D., Harte, N., Lopez, R., & Apweiler, R. (2004). The Gene Ontology Annotation (GOA) Database: Sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Research, 32(Database issue), D262-D266. https://doi.org/10.1093/nar/gkh021
Caplan, P. (2003). Metadata fundamentals for all librarians. American Library Association.
Carroll, J.M. (1997). Scenario-based design. In M. Helander & T.K. Landauer (Eds.). Handbook of human–computer interaction. 2nd ed. (pp. 383–406). North Holland.
Caufield, J. H., Zhou, Y., Garlid, A. O., Setty, S. P., Liem, D. A., Cao, Q., Lee, J. M., Murali, S., Spendlove, S., Wang, W., Zhang, L., Sun, Y., Bui, A., Hermjakob, H., Watson, K. E., & Ping, P. (2018). A reference set of curated biomedical data and metadata from clinical case reports. Scientific Data, 5(1), 1-18. https://doi.org/10.1038/sdata.2018.258
Chen, E. S., & Sarkar, I. N. (2011). Towards structuring unstructured GenBank metadata for enhancing comparative biological studies. AMIA Summits on Translational Science Proceedings, 2011, 6-11. Internet Archive
Chervitz, S. A., Deutsch, E. W., Field, D., Parkinson, H., Quackenbush, J., Rocca-Serra, P., Sansone, S.-A., Stoeckert Jr., C. J., Taylor, C. F., Taylor, R., & Ball, C. A. (2011). Data standards for Omics data: the basis of data sharing and reuse. In Bioinformatics for Omics data (pp. 31-69). Humana Press. https://doi.org/10.1007/978-1-61779-027-0_2
Cochrane, G., Akhtar, R., Aldebert, P., Althorpe, N., Baldwin, A., Bates, K., Bhattacharyya, S., Bonfield, J., Bower, L., & Browne, P. (2008). Priorities for nucleotide trace, sequence and annotation data capture at the Ensembl Trace Archive and the EMBL Nucleotide Sequence Database. Nucleic Acids Research, D5-D12. https://doi.org/10.1093/nar/gkm1018
Corpas, M., Kovalevskaya, N. V., McMurray, A., & Nielsen, F. G. (2018). A FAIR guide for data providers to maximise sharing of human genomic data. PLoS computational biology, 14(3), e1005873. https://doi.org/10.1371/journal.pcbi.1005873
Crasto, C., Marenco, L., Miller, P., & Shepherd, G. (2002). Olfactory Receptor Database: a metadata-driven automated population from sources of gene and protein sequences. Nucleic Acids Research, 30(1), 354-360. https://doi.org/10.1093/nar/30.1.354
Crick, F. (1970). Central dogma of molecular biology. Nature, 227(5258), 561-563. Internet Archive
Crystal, A., & Greenberg, J. (2005). Usability of a metadata creation application for resource authors. Library & Information Science Research, 27(2), 177-189. https://doi.org/10.1016/j.lisr.2005.01.012
Davidson, S., Cohen-Boulakia, S., Eyal, A., Ludäscher, B., McPhillips, T., Bowers, S., Anand, M. K., & Freire, J. (2007). Provenance in scientific workflow systems. Bulletin of the IEEE Computer Society Technical Committee on Data Engineering, 30(4), 44-50. https://core.ac.uk/download/pdf/276286537.pdf. Internet archive
Deck, J., Gaither, M. R., Ewing, R., Bird, C. E., Davies, N., Meyer, C., Riginos, C., Toonen, R. J., & Crandall, E. D. (2017). The Genomic Observatories Metadatabase (GeOMe): a new repository for field and sampling event metadata associated with genetic samples. PLoS biology, 15(8), e2002925. https://doi.org/10.1371/journal.pbio.2002925
Ding, L., Wendl, M. C., Koboldt, D. C., & Mardis, E. R. (2010). Analysis of next generation genomic data in cancer: accomplishments and challenges. Human Molecular Genetics, 19 (2), 188-196. https://doi.org/10.1093/hmg/ddq391
Dugan, V.G., Emrich, S.J., Giraldo-Calderón, G.I., Harb, O.S., Newman, R.M., Pickett, B.E., Schriml, L.M., Stockwell, T.B., Stoeckert Jr., C.J., Sullivan, D.E., Singh, I., Ward, D.V., Yao, A., Zheng, J., Barrett, T., Birren, B., Brinkac, L., Bruno, V.M., Caler, E. … Scheuermann, R.H. (2014) Standardized metadata for human pathogen/vector genomic sequences. PLoS ONE, 9(6): e99979. https://doi.org/10.1371/journal.pone.0099979
Duval, E., Hodgins, W., Sutton, S., & Weibel, S. L. (2002). Metadata principles and practicalities. D-lib Magazine, 8(4), 16, 1-10. https://doi.org/10.1045/april2002-weibel
Field, D., Garrity, G., Gray, T., Morrison, N., Selengut, J., Sterk, P., Tatusova, T., Thomson, N., Allen, M. J., Angiuoli, S. V., Ashburner, M., Axelrod, N., Baldauf, S., Ballard, S., Boore, J., Cochrane, G., Cole, J., Dawyndt, P., De Vos, P., dePamphilis, C., Edwards, R., Faruque, N., Feldman, R., Gilbert, J., ... Wipat, A. (2008). The minimum information about a genome sequence (MIGS) specification. Nature Biotechnology, 26(5), 541. https://doi.org/10.1038/1360
Garshol, L. M. (2004). Metadata? Thesauri? Taxonomies? Topic maps! Making sense of it all. Journal of information science, 30(4), 378-391. https://doi.org/10.1177/0165551504045856
Go, K., & Carroll, J. (2004). Scenario-based task analysis. In D. Diaper & N. Stanton (Eds.). The handbook of task analysis for human-computer interaction. (pp. 117–133). Erlbaum.
Go, K., & Carroll, J. M. (2004). The blind men and the elephant: views of scenario-based system design. Interactions, 11(6), 44-53. https://doi.org/10.1145/1029036.1029037
Goll, J., Montgomery, R., Brinkac, L. M., Schobel, S., Harkins, D. M., Sebastian, Y., Shrivastava, S., Durkin, S., & Sutton, G. (2010). The Protein Naming Utility: a rules database for protein nomenclature. Nucleic Acids Research, 38(suppl_1), D336-D339. https://doi.org/10.1093/nar/gkp958
Greenberg J., Spurgin K., & Crystal A. (2006). Functionalities for automatic metadata generation applications: a survey of metadata experts’ opinions. International Journal of Metadata, Semantics and Ontologies, 1(1), 3-20. https://doi.org/10.1504/IJMSO.2006.008766
Greenberg, J. (2005). Understanding metadata and metadata schemes. Cataloging & Classification Quarterly, 40(3-4), 17-36. https://doi.org/10.1300/J104v40n03_02
Greenberg, J. (2010). Metadata for scientific data: historical considerations, current practice, and prospects. Journal of Library Metadata, 10(2-3), 75-78. https://doi.org/10.1080/19386389.2010.520262
Greenberg, J., & Robertson, W. D. (2002, October). Semantic Web construction: an inquiry of authors’ views on collaborative metadata generation. In International Conference on Dublin Core and Metadata Applications, (pp. 45-52). Florence, Italy. Internet Archive
Greenberg, J., Pattuelli, M. C., Parsia, B., & Robertson, W. D. (2001, October). Author-generated Dublin Core metadata for web resources: a baseline study in an organization. In International Conference on Dublin Core and Metadata Applications (pp. 38-45). Florence, Italy. Internet Archive
Griffin, P. C., Khadake, J., LeMay, K. S., Lewis, S. E., Orchard, S., Pask, A., Pope, B., Roessner, U., Russell, K., Seemann, T., Treloar, A., Tyagi, S., Christiansen, J. H., Dayalan, S., Gladman, S., Hangartner, S. B., Hayden, H. L., Ho, W. W. H., Keeble-Gagnère, G., Korhonen, P. K., Neish, P., Prestes, P. R., Richardson, M. F., Watson-Haigh, N. S., Wyres, K. L., Young, N. D., & Schneider, M. V. (2018) Best practice data life cycle approaches for the life sciences. F1000Research, 6, 1618, 1-28. https//doi.org/10.12688/f1000research.12344.2
Hankeln, W., Wendel, N. J., Gerken, J., Waldmann, J., Buttigieg, P. L., Kostadinov, I., Kottmann, R., Yilmaz, P., & Glöckner, F. O. (2011). CDinFusion–Submission-Ready, On-Line Integration of Sequence and Contextual Data. PloS one, 6(9), e24797. https://doi.org/10.1371/journal.pone.0024797
Hong, E. L., Sloan, C. A., Chan, E. T., Davidson, J. M., Malladi, V. S., Strattan, J. S., Hitz, B. C., Gabdank, I., Narayanan, A. K., Ho, M., Lee, B. T., Rowe, L. D., Dreszer, T. R., Roe, G. R., Podduturi, N. R., Tanaka, F., Hilton, J. A., & Cherry, J. M. (2016). Principles of metadata organization at the ENCODE data coordination center. Database, 2016, baw001. https://doi.org/10.1093/database/baw001
Huang, H. (2018). Big data to knowledge–Harnessing semiotic relationships of data quality and skills in genome curation work. Journal of Information Science, 44(6), 785-801. https://doi.org/10.1177/0165551517748291
Huang, H., Jörgensen, C. (2013). Characterizing user tagging and co-occurring metadata in general and specialized metadata collections. Journal of the American Society for Information Science and Technology, 64(9), 1878–1889. https://doi.org/10.1002/asi.22891
Huang, H., Jörgensen, C., & Stvilia, B. (2015). Genomics data curation roles, skills and perception of data quality. Library & Information Science Research, 37(1), 10-20. https://doi.org/10.1016/j.lisr.2014.08.003
International Society for Biocuration. (2018). Biocuration: distilling data into knowledge. PLoS biology, 16(4), e2002846. https//doi.org/10.1371/journal.pbio.2002846
Jupp, S., Malone, J., Bolleman, J., Brandizi, M., Davies, M., Garcia, L., Gaulton, A., Gehant, S., Laibe, C., Redaschi, N., Wimalaratne, S. M., Martin, M., Le Novère, N., Parkinson, H., Birney, E., & Jenkinson, A. M. (2014) The EBI RDF platform: linked open data for the life sciences. Bioinformatics, 30(9), 1338-1339. https://doi.org/10.1093/bioinformatics/btt765
Khatri, P., Sellamuthu, S., Malhotra, P., Amin, K., Done, A., & Draghici, S. (2005). Recent additions and improvements to the Onto-Tools. Nucleic Acids Research, 33(suppl_2), W762-W765. https://doi.org/10.1093/nar/gki472
Khatri, V., & Brown, C. V. (2010). Designing data governance. Communications of the ACM, 53(1), 148-152. https://doi.org/10.1145/1629175.1629210
Klimke, W., O'Donovan, C., White, O., Brister, J. R., Clark, K., Fedorov, B., Mizrachi, I., Pruitt, K. D., & Tatusova, T. (2011). Solving the problem: genome annotation standards before the data deluge. Standards in genomic sciences, 5(1), 168-193. https://doi.org/10.4056/sigs.2084864
Kottmann, R., Gray, T., Murphy, S., Kagan, L., Kravitz, S., Lombardot, T., Field, D., & Glöckner, F. O. (2008). A standard MIGS/MIMS compliant XML schema: toward the development of the Genomic Contextual Data Markup Language (GCDML). OMICS A Journal of Integrative Biology, 12(2), 115-121. https://doi.org/10.1089/omi.2008.0A10
Larsson, O., & Sandberg, R. (2006). Lack of correct data format and comparability limits future integrative microarray research. Nature biotechnology, 24(11), 1322-1323. https://doi.org/10.1038/nbt1106-1322
Lee, J. S. H., Kibbe, W. A., & Grossman, R. L. (2018). Data harmonization for a molecularly driven health system. Cell, 174(5), 1045-1048. https://doi.org/10.1016/j.cell.2018.08.012
Liolios, K., Chen, I. M. A., Mavromatis, K., Tavernarakis, N., Hugenholtz, P., Markowitz, V. M., & Kyrpides, N. C. (2010). The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic acids research, 38(suppl 1), D346-D354. https://doi.org/10.1093/nar/gkp848
Miles, M. B., & Huberman, A. M. (1994). Qualitative data analysis: an expanded sourcebook. (2nd ed.). Sage Publications.
Miller, E. J. (2001). An introduction to the resource description framework. Journal of library administration, 34(3-4), 245-255. https://doi.org/10.1300/J111v34n03_04
Oliver, S. G. (2006). From genomes to systems: the path with yeast. Philosophical Transactions of the Royal Society B: Biological Sciences,361(1467), 477-482. https://doi.org/10.1098/rstb.2005.1805
Ouzounis, C. A., & Karp, P. D. (2002). The past, present and future of genome-wide re-annotation. Genome Biology, 3(2), c2001. https://doi.org/10.1186/gb-2002-3-2-comment2001
Peters, B., & OBI Consortium. (2009). Ontology for biomedical investigations. Nature Precedings, 1-1. Internet Archive
Pruitt, K.D., Tatusova, T., Brown G.R., & Maglott, D.R. (2012). NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Research, 40(D1), D130-D135. https://doi.org/10.1093/nar/gkr1079
Qin, J., & Li, K. (2013). How portable are the metadata standards for scientific data? A proposal for a metadata infrastructure. In Thirteenth International Conference on Dublin Core and Metadata Applications, Lisbon, Portugal, September 2-6, 2013. (pp. 25-34) [Publisher?] https://doi.org/10.23106/dcmi.952136097
Qin, J., Ball, A., & Greenberg, J. (2012). Functional and architectural requirements for metadata: supporting discovery and management of scientific data. In Twelfth International Conference on Dublin Core and Metadata Applications, Kuching, Malaysia, September 3-7, 2012. (pp. 62-71) https://dcpapers.dublincore.org/article/952135997
Rapp, B. A., & Wheeler, D. L. (2005). Bioinformatics resources from the National Center for Biotechnology Information: an integrated foundation for discovery. Journal of the American Society for Information Science and Technology, 56(5), 538-550. https://doi.org/10.1002/asi.20142
Samuel, V., Gussman, A., & Klumke, W. (2008). Toward an online repository of standard operating procedures (SOPs) for (meta)genomic annotation. OMICS: A Journal of Integrative Biology, 12(2), 137–141. https://doi.org/10.1089/omi.2008.0017
Schadt, E. E., Linderman, M. D., Sorenson, J., Lee, L., & Nolan, G. P. (2010). Computational solutions to large-scale data management and analysis. Nature Reviews Genetics, 11(9), 647-657. https://doi.org/10.1038/nrg2857
Singh, G., Bharathi, S., Chervenak, A., Deelman, E., Kesselman, C., Manohar, M., Patil, S., & Pearlman, L. (2003, November). A metadata catalog service for data intensive applications. In Supercomputing, 2003 ACM/IEEE Conference (pp. 33-33). IEEE. https://doi.org/10.1145/1048935.1050184
Smith, B., Ashburner, M., Rosse, C., Bard, J., Bug, W., Ceusters, W., Goldberg, L. J., Eilbeck, K., Ireland, A., Mungall, C. J., The OBI Consortium, Leontis, N., Rocca-Serra, P., Ruttenberg, A., Sansone, S.-A., Scheuermann, R. H., Shah, N., Whetzel, P. L., & Lewis, S. (2007).The OBO Foundry: coordinated evolution of ontologies to support biomedical data integration. Nature biotechnology, 25(11), 1251-1255. https://doi.org/10.1038/nbt1346
Stathias, V., Koleti, A., Vidović, D., Cooper, D. J., Jagodnik, K. M., Terryn, R., Forlin, M., Chung, C., Torre, D., Ayad, N., Medvedovic, M., Ma'ayan, A., Pillai, A., & Schürer, S. C. (2018). Sustainable data and metadata management at the BD2K-LINCS Data Coordination and Integration Center. Scientific data, 5, 180117. https://doi.org/10.1038/sdata.2018.117
Stein, L. (2001). Genome annotation: From sequence to biology. Nature Reviews Genetics, 2, 493–503. https://doi.org/10.1038/35080529
Tan, T.W., Tong, J.C., De Silva, M., Lim, K.S., & Ranganathan, S. (2010). Advancing standards for bioinformatics activities: persistence, reproducibility, disambiguation and minimum information about a bioinformatics investigation (MIABi). BMC Genomics, 11, Suppl. 4, S27. https://doi.org/10.1186/1471-2164-11-S4-S27
Vasilevsky, N., Johnson, T., Corday, K., Torniai, C., Brush, M., Segerdell, E., Wilson, M., Shaffer, C., Robinson, D., & Haendel, M. (2012). Research resources: curating the new eagle-i discovery system. Database, 2012, bar067. https://doi.org/10.1093/database/bar067
Whetzel, P. L., Noy, N. F., Shah, N. H., Alexander, P. R., Nyulas, C., Tudorache, T., & Musen, M. A. (2011). BioPortal: enhanced functionality via new Web services from the National Center for Biomedical Ontology to access and use ontologies in software applications. Nucleic Acids Research, 39 (Web Server issue), W541–W545. https://doi.org/10.1093/nar/gkr469.
Wieczorek, J., Bloom, D., Guralnick, R., Blum, S., Döring, M., Giovanni, R., Robertson, T., & Vieglais, D. (2012). Darwin Core: an evolving community-developed biodiversity data standard. PloS one, 7(1), e29715. https://doi.org/10.1371/journal.pone.0029715
Wilkinson, M. D., Dumontier, M., Aalbersberg, I. J., Appleton, G., Axton, M., Baak, A., Blomberg, N., Boiten, J.-W., Bonino da Silva Santos, L., Bourne, P. E., Bouwman, J., Brookes, A. J., Clark, T., Crosas, M., Dillo, I., Dumon, O., Edmunds, S., Evelo, C. T., Finkers, R., Gonzalez-Beltran, A., Gray, A. J. G., Groth, P., Goble, C., Grethe, J. S., ... Mons, B. (2016). The FAIR Guiding Principles for scientific data management and stewardship. Scientific data, 3. Article 160018. https://doi.org/10.1038/sdata.2016.18
Willis C., Greenberg J., & White H. (2012). Analysis and synthesis of metadata goals for scientific data. Journal of the American Society for Information Science and Technology, 63, 1505-1520. https://doi.org/10.1002/asi.22683.
Yilmaz, P., Kottmann, R., Field, D., Knight, R., Cole, J. R., Amaral-Zettler, L., Glockner, F. O. (2011). Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nature Biotechnology, 29, 415–420. https://doi.org/10.1038/nbt.1823
Zappa, A., Splendiani, A., & Romano, P. (2012). Towards linked open gene mutations data. BMC bioinformatics, 13(Suppl 4), S7.
Zeng, M.L. & Qin, J. (2022). Metadata. 3rd ed. ALA-Neal-Schuman.